
background and overview[1]
ethoxyformylmethyltriphenylphosphine bromide is an organic phosphine compound that can be used as a pharmaceutical synthesis intermediate.
preparation[1]
ethoxyformylmethyl triphenylphosphine bromide is prepared as follows: add ethyl acetate (5.0 l) to triphenylphosphonium, and add ethyl bromoacetate (665 g, 4.01 mol) dropwise to the mixture while stirring. , after the dropwise addition, the reaction mixture was stirred at room temperature for 24 hours. the mixture was then suction filtered, and the filter cake was washed three times with 500 ml of ethyl acetate each time. the filter cake is vacuum dried to obtain ethoxyformylmethyltriphenylphosphine bromide [ph3pch2co2 et]+br- (1500 g) white solid, yield 92%.
apply[1]
ethoxyformylmethyltriphenylphosphine bromide can be used as a pharmaceutical synthesis intermediate. if the following reaction occurs:
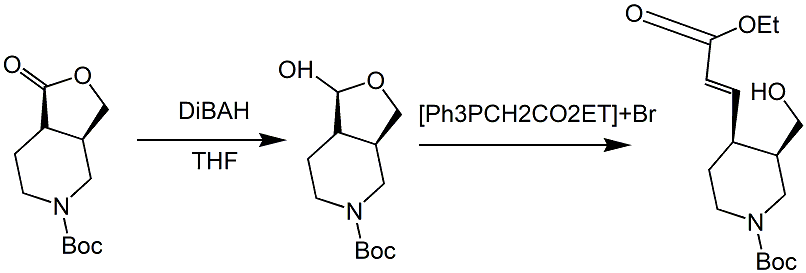
1) add tetrahydrofuran (1.5 l) to compound 8a (103 g, 0.427 mol), stir and control the temperature at -70°c, add dibah (855 ml, 0.855 mol, 1m toluene solution) dropwise into the reaction solution, and add it dropwise after the addition was complete, the reaction mixture was stirred under nitrogen at -70°c for 1.5 hours. tlc (volume ratio of petroleum ether/ethyl acetate 2:1) showed that the reaction was complete, and the reaction mixture was quenched with methanol (500 ml) and stirred at room temperature for 30 minutes. then the funnel was padded with diatomaceous earth and the reaction mixture was suction-filtered, and the filter cake was washed with a solution with a dichloromethane/methanol volume ratio of 20/1 until the product was completely washed out of the filter cake. the filtrate was concentrated under reduced pressure, and the final residue was purified by silica gel column chromatography (gradient elution: dichloromethane/methanol volume ratio from 200/1~100/1 to 50/1 to 20/1) to obtain compound 9 as a yellow oil ( 67 g), yield 64%.
2) add dichloromethane (2.0 l) to ethoxyformylmethyltriphenylphosphine bromide (706.6 g, 1.646 mol), and add sodium hydroxide (98.7 g, 2.469 mol) dropwise while the mixture is stirred. and water (500 ml). after the dropwise addition was completed, the mixture was stirred at room temperature for 10 minutes. the organic phase was then separated and the aqueous phase was extracted twice with 200 ml of methylene chloride each time. the combined organic phases were dried over anhydrous sodium sulfate and concentrated under reduced pressure. the concentrated residue was added to a mixture of compound 9 (200 g, 0.823 mol) and toluene (1.0 l), and the final reaction mixture was stirred at 60 °c for 5 h. tlc (the volume ratio of petroleum ether/ethyl acetate was 2:1) showed that the reaction was completed. the reaction mixture was concentrated under reduced pressure to obtain yellow oily impure compound 10 (800g), with a yield greater than 100%.
main reference materials
[1] cn107383035 – preparation method of 2-((1s3ar7ar)-5-tert-butoxycarbonyltetrahydrofuro[3,4]piperidine-1)acetic acid
 微信扫一扫打赏
微信扫一扫打赏

