
Background and overview[1][2]
(R)-2-(Aminoethyl)-4-chlorophenol is a chiral organic intermediate, which can be prepared by reductive amination of 2-hydroxy-5-chloroacetophenone and then chiral separation. There are reports in the literature that (R)-2-(aminoethyl)-4-chlorophenol is used to prepare chiral phosphine oxide compounds.
Application examples[2]
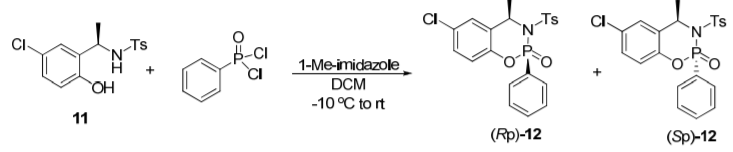
(R)-2-(Aminoethyl)-4-chlorophenol can be used to prepare a chiral phosphine oxide compound. Chiral phosphine oxides have been demonstrated to exhibit excellent enantioselectivity in transition metal-catalyzed asymmetric reactions. As shown in the figure below, (R)-2-(aminoethyl)-4-chlorophenol can be prepared through a three-step reaction to (Rp)-12. (Rp)-12 can be used to prepare a variety of chiral Buchwald ligands.
The specific preparation method is as follows:
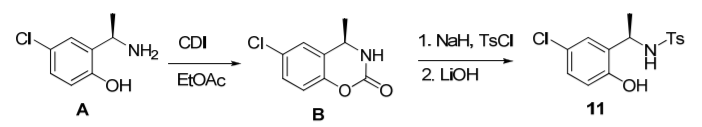
To a solution of (R)-2-(aminoethyl)-4-chlorophenol (200 g, 1.16 mol) in ethyl acetate (1600 mL) at ambient temperature was added 1,1′-carbonyldiimidazole (CDI) (200g, 1.23mol, 1.1 equivalent), and the mixture was stirred for 3-4 hours to complete the reaction. The slurry is filtered to obtain the first batch of product. Add HCl (1.0L, 1.25M) to the filtrate, stir for 15 minutes, and remove the water layer. The organic phase was then washed with brine (200 mL) and concentrated to approximately 200 mL. Add hexane, stir, and slurry to obtain the desired product. The products were combined and dried under vacuum to give B (223 g) in 97% yield.
Cool a slurry of NaH (30.5 g, 60%, 0.76 mol, 1.5 equiv) in THF (150 mL) to 0 °C. A mixture of B (100 g, 0.51 mol) and TsCl (100 g, 0.53 mol, 1.04 equiv) in THF (900 mL) was then added over 1.5-2 h while maintaining the temperature at <10 °C. After addition, the mixture was stirred at 0°C for 30 minutes, then heated to ambient temperature and stirred for 1-2 hours to complete the reaction. Add saturated aqueous ammonium chloride solution (150 mL), followed by water (100 mL) and ethyl acetate (500 mL), and stir. The aqueous phase was removed and the organic phase was washed with brine (100 mL). The aqueous phase was extracted with ethyl acetate (100 mL). The organic phases were combined and concentrated to remove organic solvent. LiOH (400 mL, 3M) was added and the mixture was heated at approximately 60°C for 3-4 hours. The mixture was cooled to ambient temperature and concentrated HCl was slowly added to pH approximately 4 and extracted twice with ethyl acetate (300 mL). The combined organic phases were concentrated and the residue was recrystallized from toluene/heptane (1:3, v/v) to give 11 (155 g) in 94% yield.
Cool a solution of 11 (100.0 g, 307.7 mmol) in anhydrous dichloromethane (1200 mL) to -10°C and add PhP(O)Cl2 (57.6 mL, 369.2 mmol, 90 wt%, 1.2 equiv) followed by addition of 1-methylimidazole (61.0 mL, 769.2 mmol, 2.5 equiv) over 30 min while maintaining the internal temperature <-5 °C under argon. The reaction mixture was stirred at -10°C to -5°C for 30-60 minutes to complete the reaction. Water (500 mL) was added to quench the reaction. The organic phase was washed with 400 mL 1N HCl and 100 mL water, and then with 200 mL saturated NaHCO3 aqueous solution. The organic portion was filtered through celite and concentrated. The residue was recrystallized using EtOAc:hexane (500 mL:1200 mL) to afford (RP)-12 (117.0 g) as a white solid in 85% yield and >99.5:0.5dr selectivity.
Main reference materials
[1]DesignandSynthesisofChiralOxathiozinoneScaffolds:EfficientSynthesisofHinderedEnantiopureSulfinamidesandSulfinylKetimines[J].AngewandteChemie,2013,125(26):6713-6717.
[2]EfficientAsymmetricSynthesisofP-ChiralPhosphineOxidesviaProperlyDesignedandActivatedBenzoxazaphosphinine-2-oxideAgents[J].JournaloftheAmericanChemicalSociety,2013,135(7):2474-2477.
 微信扫一扫打赏
微信扫一扫打赏

