
Background and overview[1]
P-Dodecylbenzene sulfonyl azide can be used as an intermediate for pharmaceutical and chemical synthesis. If p-dodecylbenzene sulfonyl azide is inhaled, move the patient to fresh air; if there is skin contact, take off contaminated clothing, wash the skin thoroughly with soap and water, and seek medical attention if you feel unwell; if In case of eye contact, separate eyelids, rinse with running water or saline, and seek medical attention immediately; if ingested, rinse mouth immediately, do not induce vomiting, and seek medical attention immediately.
Structure
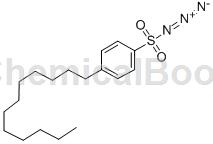
Apply [1]
P-Dodecylbenzene sulfonyl azide can be used as an intermediate for pharmaceutical and chemical synthesis. Such as the synthesis of meropenem intermediates. Meropenem was developed by Sumitomo Corporation of Japan and launched on the market in 1994. Meropenem was developed by Merck of the United States and launched in 2002. Biapenem was developed by American Cyanamid Company and launched in 2002. Doripenem was developed by Japan’s Shionogi Company and launched in 2005. Azocyclic C compounds are important intermediates of penem drugs. The structural formula of the compound of formula C is as follows:
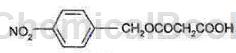
The preparation process is as follows: add 30g mono-p-nitrobenzyl malonate, 200ml methylene chloride, and 12g anhydrous magnesium chloride into the reaction bottle. At -5-40°C, add 25 ml of diisopropylethylamine dropwise, and then stir for 1-3 hours. Backup solution. Add 16g of compound A and 200ml of methylene chloride to another reaction bottle, add 12g of carbonyldiimidazole at 0-25°C, stir for 0.5-2h, and set aside the solution. The above two solutions were reacted at 30-45°C for 5-20 hours until the reaction was completed as tracked by TLC. Stir and wash the layers with hydrochloric acid, potassium carbonate aqueous solution and water respectively to obtain the organic layer of compound B. Add 8g of triethylamine and 30g of p-dodecylbenzenesulfonyl azide to the organic layer of compound B, and incubate the reaction at 0-30°C for 3 hours. After the reaction, , concentrate under reduced pressure to dryness at 25-40°C, add 320 ml of hexane, and cool to -10-5°C for crystallization for 3 hours. After suction filtration and vacuum drying at 30-50°C, 18g of compound C was obtained.
Main reference materials
[1] CN201310122884.3 Preparation method of nitrogen heterocyclic compounds
 微信扫一扫打赏
微信扫一扫打赏

