
[Overview]
N-{3-Chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-iodoquinazolin-4-amine is an important intermediate in the synthesis of lapatinib.
[Preparation method]
1. Use 2-aminobenzonitrile as raw material, iodide to obtain 2-amino-5-iodobenzonitrile, and then condense it with N,N-dimethylformamide dimethyl acetal (DMF?DMA) N’-(2-cyano-4-iodophenyl)-N,N-dimethylformamidine. Excess DMF?DMA was evaporated under reduced pressure, and glacial acetic acid and 3-chloro-4-[(3-fluorobenzyl)oxy]aniline were added to perform Dimroth rearrangement to obtain N-{3-chloro-4-[(3- Fluorobenzyl)oxy]phenyl}-6-iodoquinazolin-4-amine, yield 69.6%. This method uses iodine chloride and hydrazine hydrate during the synthesis process, which causes greater environmental pollution and a relatively low yield. 2. Using 2-amino-5-iodobenzoic acid as the starting material, the key intermediate of the new quinazoline anti-tumor drug lapatinib – N-{3-chloro- 4-[(3-fluorobenzyl)oxy]phenyl}-6-iodoquinazolin-4-amine, the total yield was 77.9%. The specific operation methods are as follows;
(1) Synthesis of 6-iodoquinazolin-4-one (3) Add 2105.2g (400mmol), 83.2g formamidine acetate (800mmol), and 1L glycerin into a three-necked flask, and heat with stirring to 120°C to completely dissolve the solid, and react at 150°C for 2 hours. Cool to room temperature, pour into 1% ammonia water (1L) with stirring, filter, wash the filter cake with water, and dry to obtain 3107.3g of off-white solid, with a yield of 99.0%.
(2) Synthesis of 4-chloro-6-iodoquinazoline (4) Add 320g (74mmol) into a three-necked flask, slowly add 350mL of SOCl2 and 3.5mL of DMF while stirring, and reflux for 2 hours. Add 200 mL of dichloromethane, evaporate excess SOCl2 under normal pressure, add 50 mL of dichloromethane to the residue, concentrate to dryness under reduced pressure, and recrystallize with ethyl acetate to obtain 420.1 g of yellow needle-like solid, with a yield of 94.4%.
(3) Synthesis of 4-(6-iodoquinazolin-4-ylamino)-2-chlorophenol (5). Add 417.28g (59mmol) and 2-chloro-4 to a three-necked flask in sequence. -Aminophenol 9.48g (65mmol), isopropyl alcohol 50mL, reflux reaction for 2 hours under stirring [TLC tracking, developing agent: V (petroleum ether): V (ethyl acetate) = 1:1, the same below]. After cooling to room temperature, the solid precipitated, filtered, and the filter cake was washed with water and dried to obtain 521.9 g of yellow-green solid, with a yield of 92.6%.
(4) Synthesis of N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-iodoquinazolin-4-amine (1) in a three-necked flask Add 5 11.9g (30mmol), acetone 250mL, m-fluorobenzyl fluoride 5.2g (36mmol), potassium carbonate 24.8g (180mmol), 18-crown-60.79g (3mmol) and KI 0.49g (3mmol) in sequence, stir under React at 35°C for 12 hours (TLC tracking). Cool to room temperature, filter, wash the filter cake fully with water, dry to obtain off-white solid, recrystallize with ethyl acetate to obtain 13.6g of yellow solid 1, yield 90.1%.
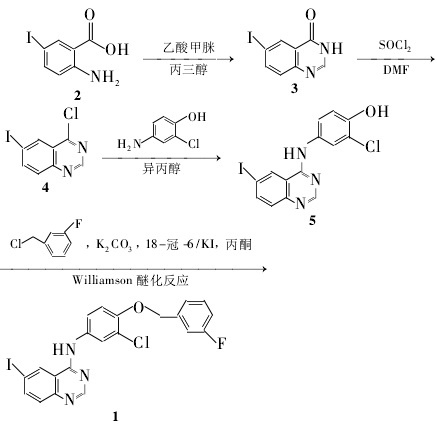
Figure 1 shows the synthetic route of method 2
[Main reference materials]
[1] Cai Zhiqiang, Shi Yu, Yuan Jing, Liu Jingguo, Liu Jinlei, Li Hongming, Huang Changjiang, Li Yiliang. N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl} Improvement of the synthesis process of -6-iodoquinazolin-4-amine [J]. Synthetic Chemistry, 2011, 19(03): 421-424.
 微信扫一扫打赏
微信扫一扫打赏

