
Background and overview[1]
1-Phenyl-2,2,2-trifluoroethylamine can be used as a pharmaceutical synthesis intermediate.
Preparation[1]
The preparation of 1-phenyl-2,2,2-trifluoroethylamine is as follows:
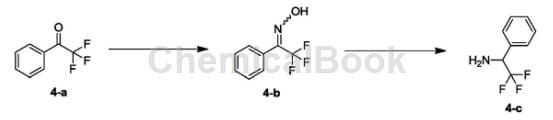
Synthesis of compound 4-b:
Dissolve 4-a (1.00g, 5.74mmol, 781.25uL, 1.00eq) in EtOH (8.00mL) and water (4.00mL) at 25°C, then add hydroxylamine hydrochloride (598.31mg, 8.61mmol, 1.50 eq) and sodium acetate (941.70 mg, 11.48 mmol, 2.00 eq). The resulting mixture was stirred at 80°C for 15 hours. The reaction was detected by TLC (PE/EA=10/1) until the reaction was complete. The reaction solution was concentrated to remove ethanol. The obtained crude product was added with water (50 mL) and stirred at 0°C for 30 minutes, filtered and concentrated to obtain white solid compound 4-b. H-NMR (400MHz, DMSO-d6) δppm7.22-7.75 (m, 5H) 12.73 (brs, 1H).
Synthesis of compound 1-phenyl-2,2,2-trifluoroethylamine: Dissolve 4-b (870.00mg, 4.59mmol, 1.00eq) in EtOH (20.00mL) and HOAc (2.00mL) Then, 10% Pd/C (87.00 mg) was added under nitrogen atmosphere. The resulting suspension was replaced three times with H2 and stirred at 25°C for 15 hours in the presence of H (50 Psi). TLC (PE/EA=3/1) detected the reaction until completion. The reaction solution was filtered and concentrated under reduced pressure. 1NNaOH (10 mL) was added to the crude product and extracted with EA (25 mL*2). The organic phases were combined and washed with saturated brine (50 mL*2), dried over NaSO4, filtered and concentrated to obtain colorless oily liquid 1-phenyl-2,2,2-trifluoroethylamine.
1HNMR (400MHz, DMSO-d6) δppm4.48 (q, J=7.91Hz, 1H) 7.26-7.62 (m, 5H).
Application
1-Phenyl-2,2,2-trifluoroethylamine can be used as a pharmaceutical synthesis intermediate. If the following reaction occurs:
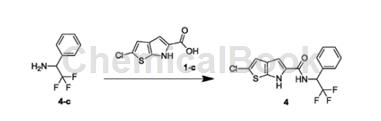
The specific steps are: combine 1-phenyl-2,2,2-trifluoroethylamine (324.99mg, 691.74umol, 1.10eq) and 1-c (150.00mg, 628.85umol, 1.00eq) at 25°C ) was dissolved in DMF (10.00mL) and then TBTU (302.87mg, 943.27umol, 1.50eq) and DIPEA (243.82mg, 1.89mmol, 329.48uL, 3.00eq) were added. The resulting mixture was stirred at 25°C for 12 hours. Reaction completion was detected by TLC (PE/EA=3/1) and LC-MS. Water (20 mL) was added to the reaction solution and extracted with EA (50 mL*3). The organic phases were combined and washed with saturated brine (30 mL*2), dried over NaSO4, filtered and concentrated to obtain the crude product. The crude product was purified by prep-HPL (Column: BostonGreenODS150*305u; mobilephase: [water (0.225% FA)-ACN]; B%: 55%-82%, 10min) to obtain compound 4.
HNMR (400MHz, DMSO-d6) δppm6.02 (quin, J=8.91Hz, 1H)7.20 (s, 1H)7.36 (s, 1H)7.39-7.50 (m, 3H)7.68 (brd, J= 6.40Hz, 2H) 8.43 (s, 1H) 9.17 (brd, J = 9.29Hz, 1H) 11.60-12.49 (m, 1H).
Main reference materials
[1]CN201810497159.7IDO inhibitor
 微信扫一扫打赏
微信扫一扫打赏

