
Background and Overview
2-Bromo-4-fluorophenyl acetonitrile is a phenylacetonitrile compound that can be used as a pharmaceutical intermediate.
Preparation[1]

Heat a solution of NaCN (2.0 equiv) in DMSO (6 mL) to 90 °C. The oil bath was removed and 2-bromo-4-fluorobenzyl bromide (1.0 equiv, 6.09 mmol) was slowly added. After the mixture reached 50 °C, water (120 mL) was added. The mixture was extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were washed with brine, dried (Na2SO 4), filtered and concentrated under reduced pressure. The crude product, 2-(2-bromo-4-fluorophenyl)acetonitrile, was purified by flash chromatography on silica gel (hexane/ethyl acetate: 9/1) as a white solid. Yield 50%, 1H NMR (400MHz, DMSO-d6): δ7.70 (dd, J = 8.6, 2.7Hz, 1H), 7.61 (dd, J = 8.6, 6.0Hz, 1H), 7.34 (dt, J = 8.6), 2.7Hz, 1H), 4.07 (s, 2H).
Apply [2]
Plk1 belongs to a small family of protein kinases characterized by a phosphoserine and threonine binding domain known as the polo box domain. Plk1 plays a central role in the regulation of the cell cycle. Among its many functions, Plk1 is thought to regulate the initiation, progression and exit of mitosis, the stages when cancer cells divide. Therefore, blocking Plk1 in cancer cells prevents cancer cells from dividing, or mitosis.
CN200880015768.9 provides novel triazoylaminopyrimidine compounds that are considered to be clinically useful in treating cancer by inhibiting Plk1. Certain of these compounds are believed to have improved efficacy compared to the compounds disclosed in WO 06/066172. The present invention provides compounds of general formula I:
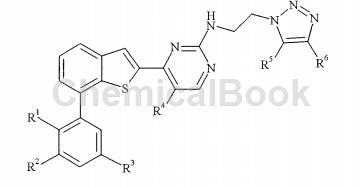
2-(2-bromo-4-fluorophenyl)-2-methylpropionitrile is an intermediate for preparing the compound of the above general formula, and 2-bromo-4-fluorophenyl acetonitrile can be used to prepare 2-(2- Bromo-4-fluorophenyl)-2-methylpropionitrile:
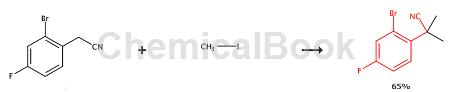
Sodium hydride (1 g, 42.1 mmol) was added to a stirred solution of 2-bromo-4-fluorobenzene acetonitrile (3 g, 14 mmol) in 10 mL dimethylformamide (DMF) at 0°C. The mixture was left at 0°C to room temperature for half an hour. Methyl iodide (6g, 42mmol) was added. Stir for another 30 minutes. Quench the reaction with water. Extract products into DCM. The organic phase was dried over sodium sulfate and concentrated to give an oily residue. The residue was purified by flash column chromatography (FCC) (hexanes to 20% ethyl acetate in hexane, gradient elution) to give the title compound as a white solid (2.2 g, 65%). 1H NMR (400MHz, CDCl3) δ7.41-7.47 (m, 2H), 7.03- 7.08 (m, 1H), 1.88 (s, 6H).
Main reference materials
[1] Kukosha, Tatyana et al, From Synlett, (17), 2525-2528; 2011
[2] CN200880015768.9 Triazoylaminopyrimidine compound
 微信扫一扫打赏
微信扫一扫打赏

