
Overview
Ethyl benzoacetate is an important raw material for the synthesis of color photographic couplers and some pharmaceutical intermediates.
Synthesis[1][2][3]
1. Acidic decomposition of ethyl formyl acetoacetate
Reaction:
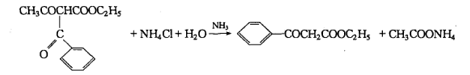
Add 6g (0.1lmol) ammonium chloride and 30mL water to a 150mL Erlenmeyer flask, then add 2mL (0.03mo1) ammonia water with a relative density of 0.9, and heat the Erlenmeyer flask in a hot water bath until 42°C, quickly add 12 g (0.05mol) of ethyl benzoyl acetoacetate at 20°C, shake the mixture, continue to heat in a hot water bath at 42°C for 10 minutes, and then cool quickly in an ice water bath. Triangular Yellow oily droplets formed at the bottom of the flask. The mixture was extracted twice with 40 mL of diethyl ether. The ether layer was separated and dried over anhydrous magnesium sulfate. The diethyl ether was evaporated in a hot water bath. Distill under reduced pressure and collect the fraction at 165°C/1862 Pa, which is ethyl benzoyl acetate. The yield is about 5 g (yield is about 52%). The pure product is a colorless and transparent liquid with a bp of 265~270℃ (partially decomposed).
2. Reaction formula:
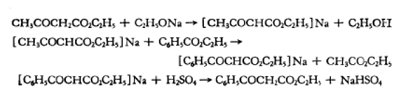
Place a 2-liter three-neck bottle equipped with a reflux condenser, a separatory funnel and an effective sealing stirrer on the steam bath, put 600 ml of absolute ethanol in the bottle, and mix 46 grams (2 grams of atoms) Cut the clean sodium into small pieces and add it in batches. After stirring and cooling the sodium ethoxide solution to room temperature, slowly add 267 grams (260 ml, 2.05 mole) of ethyl acetoacetate through a separatory funnel. Then remove the reflux condenser, install a short distillation head, and evaporate the ethanol under reduced pressure from the water pump when it is close to room temperature.
After about half of the ethanol was evaporated, a considerable amount of ethyl acetoacetate sodium salt precipitated, making stirring impossible. After the residue is evaporated to dryness (about 2 hours), it is heated on a steam bath for 1 hour under a pressure of 2 mm to remove the last remaining ethanol. The flask was allowed to cool to room temperature under reduced pressure.
Add 600 grams (570 ml, 4 moles) of ethyl benzoate to the cooled residue of ethyl acetoacetate sodium salt. Change the steam bath to an oil bath, raise the bath temperature to 140-150°C, and keep it warm for 6 hours. Then gradually increase the bath temperature to 180°C within 1 hour. The distillate collected during the heating process is 200-210 grams, the main components are ethyl acetate and ethanol.
After the reaction mixture is cooled, add 250 ml of water and a cold solution of 100 g of concentrated sulfuric acid and 200 ml of water to make the mixture acidic to litmus. Keep the mixture in a cooling state, add some crushed ice if necessary, separate the upper ester layer, extract the water layer with 200 ml of diethyl ether, combine the diethyl ether extract and the ester layer, add 350 ml of saturated sodium bicarbonate solution and shake until inert. Carbon dioxide is evolved, and the organic layer is washed with 200 ml of water.
After combining the aqueous layer and sodium bicarbonate solution, extract with 400 ml of diethyl ether. The ether layer and ester layer were combined and dried over anhydrous sodium sulfate. Evaporate the ether on a steam bath, and then evaporate excess ethyl benzoate and ethyl acetoacetate through a 15 cm fractionating column under reduced pressure. Finally, the ethyl benzoyl acetate was evaporated from 10 l to 1 06 o (1 mm). The boiling point interval is 5oThe desired yield is 190-210 grams (calculated as 50-55% of the theoretical yield based on ethyl acetoacetate).
3. In a 250 ml three-necked flask equipped with a high-speed stirrer, add 40 ml of water, 20 ml of petroleum ether (boiling range 60-90°C) and 13 g (0.1 mol) of newly distilled acetoacetic acid. ethyl ester, cool the mixture to 5°C in an ice-water bath, and then slowly add 45 ml of 33% sodium hydroxide solution while stirring. Install a 60 ml dropping funnel on the side of the three-necked flask, and put 21 ml of 33% sodium hydroxide solution into the dropping funnel; install a “Y” shaped tube on the other side of the three-necked flask, with the tube openings respectively. Equip a 60 ml dropping funnel (containing 16 grams of newly distilled benzoyl chloride, 0.11 mole) and a thermometer (the mercury ball is required to be in the liquid).
Under high-speed stirring conditions, slowly drop benzoyl chloride and sodium hydroxide solutions from two dropping funnels at the same time. During the dropwise addition, the temperature of the reaction system should be maintained below 10°C, and the pH of the reaction solution should be maintained at around 1.1. The addition time should be about half an hour. When the addition is completed, remove the ice water bath, stir at room temperature for one hour, and continue to raise the temperature. Stir to 35°C for one hour. Stop stirring, separate the water layer in the separatory funnel, and put the water layer into a 500 ml Erlenmeyer flask. Add a total of 5.5 grams of ammonium chloride in batches, stir while adding, and leave it for two days after the addition. Complete hydrolysis.
Add 6 grams of sodium chloride to the above reaction solution for salting out, separate the organic phase, extract the aqueous layer with 20 ml of benzene, combine the benzene layer and the organic phase, then wash with water twice, and then evaporate at normal pressure Benzene is removed, followed by vacuum distillation, and the fraction at 140-145°C/12 mmHg is collected. The yield of ethyl benzoacetate is approximately 60%. The pure product is a colorless and transparent liquid with a boiling point of 265-270°C (partial decomposition) and 147-159°C/11 mm Hg.
References
[1] Editor, School of Chemistry and Chemical Engineering, Lanzhou University, University Chemistry Experiment Basic Chemistry Experiment 1, Lanzhou University Press, 2004.08, page 313
[2] E.C. Horning, Organic Synthesis (Episode 3), Science Press, 1981.08, page 234
[3] Zeng Zhaoqiong, Organic Chemistry Experiments, Higher Education Press, 1981.05, page 155
 微信扫一扫打赏
微信扫一扫打赏

