
Background and overview[1]
1-Chloro-4-(4-phenyl-1,3-butadiene)benzene can be used as a pharmaceutical synthesis intermediate.
Preparation[1]
1) Synthesis of bisethyl-4-chlorobenzylphosphine
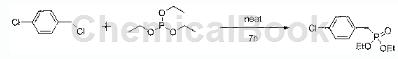
Preparation process: Preparation of bisethyl-4-chlorobenzylphosphine: 1-chloro-4-(chloromethyl)benzene (50g, 0.31mol) and triethyl phosphite (74.9g, 0.47mol) Add to a 200 ml flask, heat to reflux and stir for 7 hours. The mixture is cooled to room temperature, and excess triethyl phosphite is distilled off under reduced pressure to obtain a crude product. The product in this step does not need to be purified and can be directly used in the next step of reaction.
2) Synthesis of 41-chloro-4-(4-phenyl-1,3-butadiene)benzene
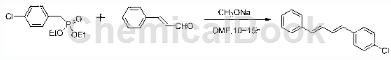
Preparation process: Add the product in 1 (39.4g, 0.15mol) to 100ml dry DMF, add 28% sodium methoxide (28.9g, 0.15mol) to the above solution, stir evenly, and add cinnamaldehyde (15.8g, 0.12mol)/DMF (10ml) solution was added to the mixture, and after stirring at room temperature for 10~15h, the mixture was poured into 300ml of water, and a small amount of HCl solution was added to obtain a large amount of white solid. Filter and use 20ml of methanol. washes. Then pour the white solid into 100ml methanol, stir, filter (twice), and dry under reduced pressure at 70°C to obtain white powder 1-chloro-4-(4-phenyl-1,3-butadiene)benzene , about 26g, yield 90%. (Melting point, characterization data).
Application
1-Chloro-4-(4-phenyl-1,3-butadiene)benzene can be used as a pharmaceutical synthesis intermediate, if the following reaction occurs:
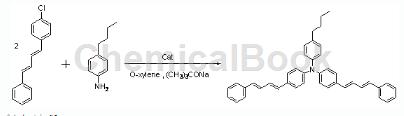
Preparation process: Accurately weigh 5.70g (0.02mol) of the product synthesized in the second step, 1-chloro-4-(4-phenyl-1,3-butadiene)benzene, in the glove box, 0.035g ( 0.1 mmol) 2-(dicyclohexylphosphino)biphenyl, 2.9 g (0.03 mol) sodium tert-butoxide and 0.022 g (0.1 mmol) palladium acetate. Under Ar gas protection, 1.49g (0.01mol) p-n-butylaniline and 150ml anhydrous and oxygen-free o-xylene were injected into the mixture, and the mixture was reacted at 130°C for 5 hours. After the reaction is completed, distill under reduced pressure at 80°C to obtain a brown liquid. Cool to 15°C overnight to obtain a brown solid; after washing with o-xylene, dissolve the solid in o-xylene at 70°C. , add silica gel, stir for 2 hours, filter while hot, and wash the silica gel with o-xylene. Collect the filtrate, distill under reduced pressure at 80°C, and cool to 15°C to obtain a yellow solid; filter, then wash with o-xylene, and finally use Wash with ethyl acetate and dry under reduced pressure at 70°C to obtain yellow-green powder. Yield 66%. Characterization: m.p.160~161°C.1HNMR (400MHz, CDCl3): 7.42 (d, 4H, J=8Hz), 7.29 (d, 8H, J=12Hz), 7.21 (d, 2H, J=8Hz), 7.08 (d, 2H, J=8Hz), 7.02 (d, 6H, J=8Hz), 6.97-6.91 (m, 2H), 6.87-6.81 (m, 2H), 6.62 (d, 2H, J=4Hz), 6.60 (d, 2H, J=4Hz), 2.58 (t, 2H, J=8Hz), 1.64-1.56 (m, 2H), 1.42-1.33 (m, 2H), 0.94 (t, 3H, J=8Hz) .
Main reference materials
[1] (CN102516088) Synthesis of a triphenylamine compound with strong fluorescence
 微信扫一扫打赏
微信扫一扫打赏

